Pathophysiology of edema in patients with chronic venous insufficiency
College of Medicine ,
Central Michigan University,
Saginaw, MI, USA
Abstract
The prevalence of chronic venous insufficiency and its associated health care costs have been greatly underestimated for a long period of time. Many patients suffering from chronic venous insufficiency either present with or have associated lower-limb edema. The main pathophysiological mechanisms of edema formation in such patients include increased capillary hydrostatic pressures secondary to valvular insufficiency or venous obstruction. In addition, there is increased capillary permeability due to associated inflammatory reactions that subsequently lead to leakage of protein-rich fluid into the interstitial space. Such fluid increases the oncotic pressure within the interstitium leading to additional edema formation. This article will review the anatomic, hemodynamic, and pathophysiologic mechanisms of lower-limb edema associated with chronic venous insufficiency. The clinical methods most commonly used to assess the extent and severity of edema formation will also be briefly examined.
Introduction
According to the VEIN-TERM transatlantic interdisciplinary consensus document published in 2009, chronic venous insufficiency (CVI) was defined as “any morphological and functional abnormalities of the venous system of long duration manifested either by symptoms and/or signs indicating the need for investigation and/or care.”1 It was further defined as applying to functional abnormalities of the venous system that resulted in producing moderate to severe edema. Therefore, CVI should be reserved for the description of more advanced disease, beginning with venous edema (C3), but more commonly in conditions where skin changes (C4) or ulceration (C5-6) have taken place, as defined in the Clinical, Etiological, Anatomical, and Pathophysiological (CEAP) classification system.2 Chronic venous disorders are one of the most prevalent, yet greatly underestimated, chronic health disorders in the USA and worldwide. A recent report by the SAGE group estimated that CVI affects approximately 175 million US citizens with an estimated cost of $46 billion annually.3,4 Worldwide, investigators from the Vein Consult Program prospectively surveyed over 90,000 individuals from different continents and found that the prevalence of symptomatic CVI was similar between Western/Eastern Europe (78%/87%), Latin America (88%), the Middle East (85%), and the Far East (87%).5
Several population-based epidemiological studies have investigated demographic and clinical factors associated with the development of CVI. Among those, advancing age is one of the most well-established risk factors for the development of CVI. In the Framingham study, Robertson reported that the risk of CVI was 1% for men and 10% for women younger than 30 years, but this increased to 57% and 77% for men and women aged 70 years or older, respectively.6 With the aging population in the USA and other Western countries, CVI incidence and prevalence are expected to increase even further in the coming years. Sex is another important risk factor. In the San Diego population-based study, Criqui reported that females were twice as likely to develop varicose veins compared with their male counterparts.7 However, in the Edinburgh vein cross-sectional population survey, which examined chronic venous insufficiency rather than just varicose veins, a higher incidence was found in males.8 Other previously reported CVI risk factors include pregnancy and a positive family history. A high incidence of CVI was also found to be associated with certain genetic disorders, such as Klippel- Trenaunay syndrome, Von Hippel-Lindau syndrome, and FOXC2 and NOTCH3 abnormalities.9-12 On the other hand, there is currently no strong evidence showing that smoking, hypertension, obesity, or decreased physical activity are associated with a higher incidence of developing CVI.
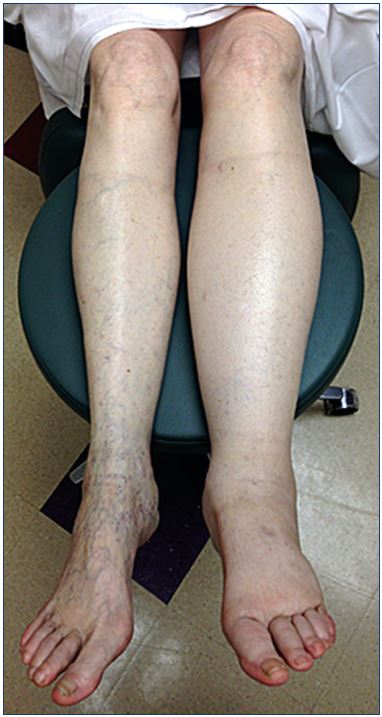
Figure 1. Patient with left leg swelling secondary to chronic
venous insufficiency due to proximal iliac vein stenosis.
Edema, defined as the accumulation of fluid in the interstitial space leading to leg swelling, is frequently the first clinical symptom or sign associated with CVI (Figure 1). A multitude of disorders, including cardiac, renal, and hepatic diseases, as well as medications, can induce leg swelling. Lymphedema, in particular, can cause severe and persistent leg edema. However, it has been estimated that CVI-associated edema represents 90% of all lower-limb edema because of the prevalence of venous insufficiency.13 The different mechanisms of lower-limb edema formation associated with the pathophysiology of CVI will herein be examined.
Anatomy and hemodynamic considerations
The anatomy of the lower extremity venous system can be highly variable among individuals, making the definition of a “normal anatomy” challenging. The International Interdisciplinary Consensus Statement in 2002 was helpful in updating the anatomical nomenclature of the venous system.14,15 The interplay between the three venous systems of the leg, the deep and superficial systems, and the connecting perforators are important in the hemodynamic changes that occur in disease states. While the deep system accompanying the major arteries of the leg return approximately 80% of the lower-limb venous blood, and the superficial system is associated with visible varicosities and are the ones most subject to therapeutic interventions, it is the more than 150 perforating veins that provide the collateral pathways for pathologic retrograde flow that can be important in the edema manifestation of CVI.16 Of these perforators, the most well described and of greatest clinical relevance have been the medial calf perforators involving the posterior tibial veins, but any group of them may be important in the treatment of specific patients.
Under normal physiologic conditions, a unidirectional and cephalad venous blood flow is maintained by a healthy system of venous valves, lower-limb muscular pump compressive action, and negative intra-abdominal and intra-thoracic pressure. During walking, contraction of calf muscles within their fascially enclosed compartments generates an ambulatory pressure gradient between the lower leg and thigh, which leads to displacement of blood in an antigravity direction toward the heart with the assistance of competent valves. This calf pump is most important because it contains the largest venous capacitance within the soleal and gastrocnemius sinusoids and generates the highest pressures. Intramuscular pressures generated in the gastrocnemius and soleus muscles can increase up to 250 mm Hg from 9 mm to 15 mm Hg in their relaxed state.17 Valvular function, in all three venous systems, is important in the maintenance of antegrade venous flow. It is most helpful that modern ultrasound technology allows us both to directly visualize the valves and quantify their function as exemplified by valve closure times. The initial studies by van Bemmelen et al established a normal value of 0.5 seconds for the valves to close in the superficial system.18 For the deep system, 1 second or more is suggestive of valvular dysfunction, while 0.35 seconds of reversed flow in combination with a diameter of greater than 3.5 mm is generally accepted as abnormal for leg perforators.19
In pathologic states, be it due to prior episodes of deep venous thrombosis or superficial thrombophlebitis, or when there is primary valvular dysfunction, venous reflux with retrograde blood flow takes place. In the deep system, after the initial thrombotic event, intrinsic thrombolysis and recanalization allows for blood flow to resume within the previously occluded vein. However, the inflammatory and fibrotic processes that take place on the valve cusp restrict the movement of the valve leaflets resulting in only a partially mobile leaflet or a completely “frozen valve.” In addition, inflammation of the nonvalvular segments of the vein can lead to thickening and calcification in the vein wall. It is unclear to what degree this loss of elasticity and distensibility affects venous flow hemodynamics, but, at a minimum, it must decrease volume flow in those segments because of the diminished luminal diameter. The result of these processes is valvular incompetence and reflux (Figure 2).20 Dysfunction in venous valves most frequently affects all three venous systems in patients with more advanced CVI, particularly those with CEAP C6 disease with active ulcers.21-23 This leads to venous hypertension, which can be quantified by elevated ambulatory pressures in the superficial venous system when measured in the pedal veins.24 In these circumstances, venous hypertension and the associated increase in hydrostatic capillary pressure leads to edema formation.
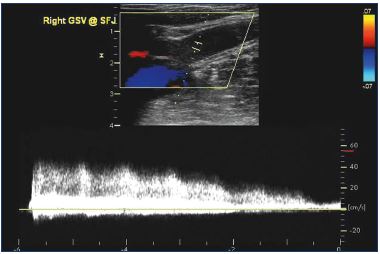
Figure 2. Longitudinal ultrasound image demonstrating six
seconds of high volume reflux at the proximal saphenofemoral
junction of the great saphenous vein.
Transcapillary fluid balance
In order to have an understanding of the pathophysiology of CVI-associated edema, one should have an appreciation of the basic physiologic principles behind transcapillary fluid exchange. Under physiologic conditions, total interstitial fluid volume is kept fairly constant, even in conditions that lead to large increases in tissue fluid transfer, secondary to the balance between transcapillary filtration and lymphatic outflow. In 1896, the British physiologist Ernest Starling was the first to describe the counteraction between hydrostatic and oncotic forces that act across the semipermeable capillary wall and controls fluid filtration from the intravascular to the extravascular space.25 Venous hypertension extends into the microcirculation with increased hydrostatic pressure in capillaries that lead to wall remodeling and microvenous valvular insufficiency.26 There is a secondary enlargement of the capillary bed, excessive transcapillary filtration causing interstitial fluid accumulation with exudation of fibrinogen and proteins into the interstitial space, which later produce the characteristic changes of lipodermatosclerosis.27 Plasma proteins, mainly albumin, are responsible for the intravascular oncotic pressure. Increased transcapillary pressures and oncotic attraction with progressive protein accumulation in the interstitial space is thought to be the primary pathophysiologic mechanism in the development of CVI-associated edema.21,28
This mechanistic understanding has been further elucidated with details of the molecular basis of this process. Almost a century after Starling, Curry and Michel explained that transcapillary fluid movement reflects an ultrafiltration process through the interpolymer spaces of the glycocalyx layer of the endothelial cells of capillary membranes.29 In addition, the underlying capillary basement membrane, composed of type IV collagen and laminin, and the adjacent extracellular matrix act as second- and third order resistance layers opposing fluid movement from the intravascular space to the interstitial space.30 Initially conceived of as a nonfunctional space, the interstitium contains an extensive extracellular matrix composed of collagen fibrils upon which glycoprotein molecules are attached in a web-like manner. Beyond the contribution of transcapillary pressure and oncotic forces, in inflammatory conditions, such as those seen in CEAP C4-6, this extracellular matrix can physiologically affect transendothelial pressure differences and thus result in increased transcapillary fluid flow with associated edema.31,32 Any derangements in lymphatic drainage would only exacerbate this problem if the terminal interstitial lymphatic capillaries had a decreased ability to clear proteins and macromolecules from this relatively hyperoncotic environment.
Capillary hydrostatic pressure
Under normal physiologic conditions, the mean hydrostatic capillary pressure is tightly regulated by the balanced changes in precapillary arteriolar and to a lesser extent postcapillary venular resistance (vasoconstriction/ vasodilation). This tight balance largely contributes to maintaining a relatively constant interstitial fluid volume by adjusting transcapillary fluid filtration rate. As arteriolar resistance is four times higher than the venular resistance, changes in venular pressure, as seen with venous outflow obstruction or venous valvular insufficiency and secondary hypertension, can have a very profound effect on transcapillary filtration.
Any of a multitude of factors leading to a decrease in venous return from the lower-limb, such as deep venous outflow obstruction, multisystem valvular incompetency, and inadequate lower-limb muscle contraction, can lead to an increase in the ambulatory venous pressure up to 60 mm to 90 mm Hg.33 With the excess blood volume in both the deep and superficial veins that is associated with venous hypertension, exacerbated by standing and worsening toward the end of the day, venous distension leads to anatomic distortion and progressive valvular incompetence in a vicious circle. As the veins become maximally distended, any further increase in venous blood volume can produce a large increase in intraluminal venous pressure. Associated with this resultant venous hypertension is the associated loss of precapillary arteriolar reflex constriction that is intended to decrease the transmission of the increased venous pressure to the capillary system.34 This venoarteriolar response (VAR) involves the arterioles constricting to reduce blood flow and normalize the arteriovenous pressure difference in the face of venous hypertension. Several studies have shown that reduction in this VAR contributes significantly in the formation of edema and correlation of CVI clinical severity.21,35
Shear stress and inflammation
Another factor that plays a role in the pathophysiology of CVI-associated edema is the reduction in shear stress, which is the tangential force of the blood flow on the endothelial lining of the blood vessel. Shear stress was found to be associated with regenerative functional and morphological changes in endothelial cells as well as release of anti-inflammatory and vasodilatory molecules.36 However, the reduction in shear stress associated with venous hypertension has been found to be associated with proinflammatory changes in the vein wall and valves. The venous wall inflammatory process leads to gap formation between endothelial cells secondary to endothelial cells’ actin/myosin filament contraction. These gaps lead to excess permeability of plasma proteins into the interstitial space, a reduction in oncotic pressure differences between the intra- and extravascular spaces and secondary edema formation.37
The combined volume effect of venous hypertension and the reduction in endothelial shear stress causes both endothelial glycocalyx damage and the release of vasoactive substances, such as chemokines, inflammatory mediators, and adhesion molecules (ICAM-1 and E-Selectin). The increased release of the ICAM-1 adhesion molecules leads to increased leukocyte adherence. Due to altered shear stress, leukocytes begin sticking to the vein wall, migrate out of the capillary, and release inflammatory mediators. These mediators trigger local inflammation, which induces further remodeling of the adjacent venous wall and valves and aggravating the venous hypertension.38 This response also includes local monocyte and macrophage recruitment with their infiltration into the vein wall and valves.39 Chronic venous hypertension increases hypoxia-inducible factors leading to increased matrix metalloproteinase (MMP) expression/activity with resultant degradation of extracellular matrix proteins. This inflammatory cascade involving both the venous system and surrounding soft tissues ultimately leads to the classic presentation of lower limb edema, lipodermatosclerosis, and leg ulcer associated with CVI.40 Whether such inflammatory responses precede or follow valvular dysfunction and venous incompetency, is not yet conclusively elucidated.21,41 The combination of these changes contribute to endothelial cell dysfunction and venous wall and valvular damage that result in venous insufficiency.37
Assessment of lower-limb edema
Characteristically, CVI-associated edema is pitting in nature and more pronounced after prolonged standing or sitting and at the end of the day. It most commonly involves the lower legs, ankle malleolar area, and less often the foot. The skin may show a whitish discoloration from stretching. In more advanced stages of CVI, leg edema is also associated with more pronounced skin color changes, thickening, and tenderness. The Venous Clinical Severity Score (VCSS) classifies venous edema by asking the patient about the extent of leg edema that is experienced: none, edema limited to the foot and ankle, edema that extends above the ankle, but below the knee, or one that extends to the knee or above.42 The most commonly used clinical method to assess lower-limb venous edema is the pitting test wherein pressure is applied by the thumb or index finger on the medial surface of the tibia.43 Severity of leg edema is described according to the pitting depth of the tissue, the time needed for the pit to resolve, and in terms of the extent to which it involves the length of the leg.44 Although easy to perform, it is an unvalidated and subjective measure of edema and may be uncomfortable for the patient. Other more objective methods that indirectly assess lower-limb edema include measuring the leg circumference at a single point or multiple fixed points using a measuring tape, usually at 3 cm above the ankle or the calf at its largest circumference. The Leg- O-Meter device utilizes a measuring tape attached to a stand, thus ensuring that measurements are taken at the same point at different times.45 Other more sophisticated methods employing mathematical computations have also been used to estimate lower-leg edema volume. These include the Frustrum method, which assumes that the lower-limb is a truncated cone and utilizes an upper and lower leg circumference as reference points from which the leg volume is calculated.46 The gold standard for measurement of lower-limb volume is a direct method utilizing volume displacement measurements wherein the leg is immersed in a water-filled container and the volume of displaced water is measured.47 Several considerations of this technique have been proposed to improve accuracy and reproducibility, including volumeter type, patient/leg/ feet positions, as well as room and water temperature.
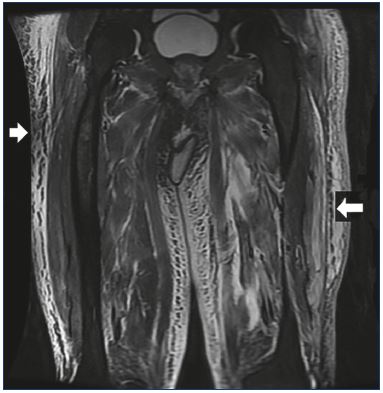
Figure 3. Coronal fat-suppressed T2-weighted magnetic
resonance image of the legs demonstrating diffuse
subcutaneous edema (white arrows).
More recently, potentially less cumbersome imaging modalities have been used to estimate lower-limb volume. In the optoelectronic measurement method, infrared rays are used to measure the lower-limb to produce a three dimensional image from which the leg volume can be electronically calculated.48 High-frequency ultrasound imaging has been used to assess dermal thickness associated with CVI edema.49 Computed tomography has been used to measure lower-limb volume and to localize edema to the different leg compartments.50 Magnetic resonance imaging has been shown to have good sensitivity in detecting venous lower-limb edema and in differentiating between lymphedema, venous edema, or lipedema (Figure 3).47 The cost, availability, and associated radiation exposure make CT and MRI less attractive modalities. Duplex ultrasound, on the other hand, has a greater appeal because of its wide availability, ease of use, and technical familiarity to phlebologists and other vascular specialists.51 Rather than just examining the skin, B-mode gray scale ultrasonography with a 15 to 18 MHz linear probe allows direct visualization of anechoic areas representing subcutaneous edema (Figure 4). Suehiro et al have proposed that subcutaneous echogenicity and subcutaneous echo-free space semi-quantitation can reflect tissue inflammation and fluid accumulation, respectively (Figure 5).52 Efforts have been made to quantify the degree of edema, both by a qualitative grading scale as well as, more easily, measuring the greatest depth from the skin which encompasses the edema.
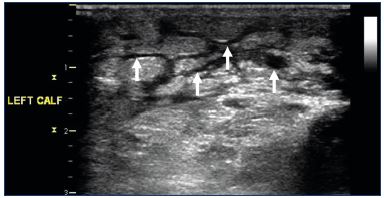
Figure 4. Transverse B-mode ultrasound image of the calf
illustrating anechoic areas of edema (arrows).
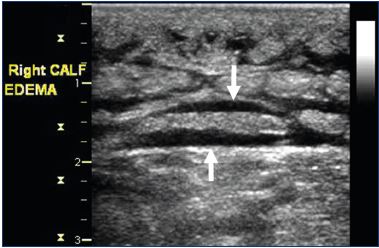
Figure 5. Longitudinal ultrasound image demonstrating
horizontally-oriented echo-free spaces (arrows) suggesting
more severe edema.
Although these qualitative and quantitative edema measurements are useful for research purposes, they have not yet been accepted for routine clinical care because of the associated additional costs and because clinical interventions have not generally utilized edema as a separate therapeutic outcome end point. However, with the increasing use of venoactive agents, which have been demonstrated to have an anti-edema effect by increasing capillary resistance, improving lymphatic drainage, and reducing capillary filtration, quantitation of leg edema may become more beneficial.53
Lower-extremity edema has long been recognized to be associated with chronic venous insufficiency. Similarly, the basic mechanism of valvular insufficiency and secondary venous hypertension leading to leg swelling and pain has been appreciated and compression therapy effectively instituted as usual clinical therapy. More recently, we have had available excellent ultrasound imaging to directly visualize valve movement and quantify reflux times as well as guiding endovascular ablation therapy. The basic hemodynamic principles of transcapillary filtration and oncotic pressure leading to edema have been known for decades. Our understanding of the details of microvascular function, molecular mediators, and inflammatory processes, however, are still at a very basic level. We have a long way to go in discovering the particulars of the intermediary pathways of edema formation and discover more specific targeted drug therapy for its control. This should provide fertile grounds for investigation in the years to come.

REFERENCES
1. Eklof B, Perrin M, Delis KT, et al. Updated terminology of chronic venous disorders: the VEIN-TERM transatlantic interdisciplinary consensus document. J Vasc Surg. 2009;49(2):498-501.
2. Eklof B, Rutherford RB, Bergan JJ, et al. Revision of the CEAP classification for chronic venous disorders: consensus statement. J. Vasc. Surg. 2004;40:1248- 1252.
3. SAGE GROUP. Chronic venous disease (CVD): epidemiology, costs and consequences. http://thesagegroup. us/pages/reports/CVD-report.php. Accessed January 8, 2020.
4. Davies AH. The seriousness of chronic venous insufficiency: a review of realworld evidence. Adv Ther. 2019;36(suppl 1):5-12.
5. Rabe E, Guex JJ, Puskas A, Scuderi A, Fernandez Quesada F. Epidemiology of chronic venous disorders in geographically diverse populations: results from the Vein Consult Program. Int Angiol. 2012;31(2):105-115.
6. Robertson L, Evans C, Fowkes FG. Epidemiology of chronic venous insufficiency. Phlebology. 2008;23:103- 111.
7. Criqui MH, Jamosmos JM, Fronek AT, et al. Chronic venous insufficiency in an ethnically diverse population. The San Diego Population study. Am J Epidemiol. 2003;158:448–56.
8. Bradbury A, Evans C, Allan P, Lee A, Ruckley CV, Fowkes FG. What are the symptoms of varicose veins? Edinburgh vein study cross sectional population survey. BMJ. 1999;318:353-356.
9. Jacob AG, Driscoll DJ, Shaughnessy WJ, Stanson AW, Clay RP, Gloviczki P. Klippel-Trenaunay syndrome: spectrum and management. Mayo Clin Proc. 1998;73:28-36.
10. Gordeuk VR, Sergueeva AI, Miasnikova GY, et al. Congenital disorder of oxygen sensing: association of the homozygous Chuvash polycythaemia VHL mutation with thrombosis and vascular abnormalities but not tumors. Blood. 2004;103:3924-3932.
11. Saiki S, Saiki K, Saiki M, et al. Varicose veins associated with CADASIL result from a novel mutation in the Notch3 gene. Neurology. 2006;67:337-339.
12. Mellor RH, Brice G, Stanton AW, et al. Mutations in FOXC2 are strongly associated with primary valve failure in veins of the lower-limb. Circulation. 2007;115(14):1912-1920.
13. Nicolaides AN. Chronic venous insufficiency and the leukocyteendothelium interaction: from symptoms to ulceration. Angiology. 2005;56(suppl 1):S11-S19.
14. Caggiati A, Bergan JJ, Gloviczki P, Jantet G, Wendell-Smith CP, Partsch H. Nomenclature of the veins of the lowerlimbs: an international interdisciplinary consensus statement. J Vasc Surg. 2002;36:416-422.
15. Kachlik D, Pechacek V, Hnatkova G, Hnatek L, Musil V, Baca V. The venous perforators of the lower-limb – a new terminology. Phlebology. 2019;34(10):650-668.
16. Van Limborgh J. L’anatomie du systemeveineux de l’extremiteinferieure en relation avec la pathologievariqueuse. Folia Angiol. 1961;8:240-257.
17. Ludbrook J. The musculovenous pumps of the human lower-limb. Am Heart J. 1966;71(5):635-641.
18. van Bemmelen PS, Bedford G, Beach K, Strandness DE. Quantitative segmental evaluation of venous valvular reflux with duplex ultrasound scanning. J. Vasc Surg. 1989;10(4):425-431.
19. Gloviczki P, Comerota AJ, Dalsing MC et al. The care of patients with varicose veins and associated chronic venous diseases: clinical practice guidelines of the Society for Vascular Surgery and the American Venous Forum. J Vasc Surg. 2011;53:2S-48S.
20. Blebea J. The pathophysiology and hemodynamics of chronic venous insufficiency. In: Glovizki P (ed). Handbook of Venous Disorders. 4th Ed. Boca Raton, FL; CRC Press; 2017:51-61.
21. Balance TF. Edema in venous insufficiency. Phlebolymphology. 2011;18(1):3-14.
22. Eberhardt RT, Raffetto JD. Chronic venous insufficiency. Circulation. 2014;130(4):333-346.
23. Thulesius O. Vein wall characteristics and valvular function in chronic venous insufficiency. Phlebology. 1993;8:94-98.
24. Reeder SW, Wolff O, Partsch H, et al. Expert consensus document on direct ambulatory venous pressure measurement. Int Angiol. 2013;32(5):453-458.
25. Starling EH. On the absorption of fluids from the connective tissue spaces. J Physiol. 1896;19:312-326.
26. Vincent JR, Jones GT, Hill GB, van Rij AM. Failure of microvenous valves in small superficial veins is a key to the skin changes of venous insufficiency. J Vasc Surg. 2011;54(suppl):62S-69S.
27. Comerota A, Lurie F. Pathogenesis of venous ulcer. Seminars Vasc Surg. 2015;28:6-14.
28. Woodcock TE, Woodcock TM. Revised Starling equation and the glycocalyx model of transvascular fluid exchange: an improved paradigm for prescribing intravenous fluid therapy. Br J Anaesth. 2012;108(3):384-394.
29. Curry FE, Michel CC. A fibre-matrix model of capillary permeability. Microvasc Res. 1980;20:96-99.
30. Levick JR. Fluid exchange across endothelium. Int J Microcirc Clin Exp. 1997;17:241-247.
31. Sarin H. Physiologic upper limits of pore size of different blood capillary types and another perspective on the dual pore theory of microvascular permeability. J Angiogenesis Res. 2010;2:14.
32. Heino J, Kapyla J. Cellular receptors of extracellular matrix molecules. Curr Pharm Design. 2009;15:1309-1317.
33. Takase S, Pascarella L, Lerond L, Bergan JJ, Schmid-Schönbein GW. Venous hypertension, inflammation and valve remodeling. Eur J Vasc Endovasc Surg. 2004;28(5):484-493.
34. Stücker M, Schöbe MC, Hoffmann K, Schultz-Ehrenburg U. Cutaneous microcirculation in skin lesions associated with chronic venous insufficiency. Dermatol Surg. 1995;21(10):877-882.
35. Labropoulos N, Wierks C, Golts E, et al. Microcirculatory changes parallel the clinical deterioration of chronic venous insufficiency. Phlebology. 2004;19(2):81- 86.
36. Li YS, Haga JH, Chien S. Molecular basis of the effects of shear stress on vascular endothelial cells. J Biomech. 2005;38(10):1949-1971.
37. Mansilha A, Sousa J. Pathophysiological mechanisms of chronic venous insufficiency and implications for venoactive drug therapy. Int J Mol Sci. 2018;19:1669.
38. Barros BS, Kakkos SK, De Maeseneer M, Nicolaides AN. Chronic venous disease: from symptoms to microcirculation. Int Angiol. 2019;38:211-218.
39. Ono T, Bergan JJ , Schmid-Schonbein GW, Takase S. Monocyte infiltration into venous valves. J Vasc Surg. 1998;27:158-166.
40. Castro-Ferreira R, Cardoso R, Leite- Moreira A, Mansilha A. The role of endothelial dysfunction and inflammation in chronic venous insufficiency. Ann Vasc Surg. 2018;46:380-393.
41. Pascarella L, Penn A, Schmid-Schönbein GW. Venous hypertension and the inflammatory cascade: major manifestations and trigger mechanisms. Angiology. 2005;56:S3-S10.
42. Vasquez MA, Rabe E, McLafferty RB, et al. Revision of the venous clinical severity score: venous outcomes consensus statement. J Vasc Surg. 2010;52:1387- 1396.
43. Cornu-Thénard A, Scuderi A, Ramelet AA, et al. UIP 2011 C3 consensus. Int Angiol. 2012;31:4-9.
44. Sinabulya H, Holmberg A, Blomgren L. Interobserver variability in the clinical assessment of the clinical severity of superficial venous insufficiency. Phlebology. 2015;30:61-65.
45. Bérard A, Kurz X, Zuccarelli F, Abenhaim L; VEINES Study Group. Venous insufficiency epidemiologic and economic study. Validity of the Leg-OMeter, an instrument to measure leg circumference. Angiology. 2002;53:21- 28.
46. Guex JJ, Perrin M. Edema and leg volume: methods of assessment. Angiology. 2000;51:9-12.
47. Rabe E, Carpentier P, Maggioli A. Understanding lower leg volume measurements used in clinical studies focused on venous leg edema. Int Angiol. 2018;37(6):437-443.
48. Engelberger RP, Blazek C, Amsler F, et al. Reproducibility and day time bias correction of optoelectronic leg volumetry: a prospective cohort study. BMC Med Res Methodol. 2011;11:138.
49. Volikova AI, Edwards J, Stacey MC, Wallace HJ. High-frequency ultrasound measurement for assessing postthrombotic syndrome and monitoring compression therapy in chronic venous disease. J Vasc Surg. 2009;50:820-825.
50. Angelhed JE, Strid L, Bergelin E, Fagerberg B. Measurement of lowerleg volume change by quantitative computed tomography. Acta Radiol. 2008;49:1024-1030.
51. Caggiati A. Ultrasonography of skin changes in legs with chronic venous disease. Eur J Vasc Endovasc Surg. 2016; 52: 534-542.
52. Suehiro K, Morikage N, Yamashita O, et al. Differentiation of functional venous insufficiency and leg lymphedema complicated by functional venous insufficiency using subcutaneous tissue ultrasonography. J Vasc Surg: Venous Lym Dis. 2017;5:96-104.
53. Nicolaides A, Kakkos S, Baekgaard N, et al. Management of chronic venous disorders of the lower-limbs. Guidelines according to scientific evidence. Part I. Int Angiol. 2018;37:181-254.